Подборка событий на выходные

Майские выходные

Отдых в Naraada за билет

Открытые катки

Гороскоп 22.04-28.04

Ваше событие на AfishaYkt

Вопросы на собесе

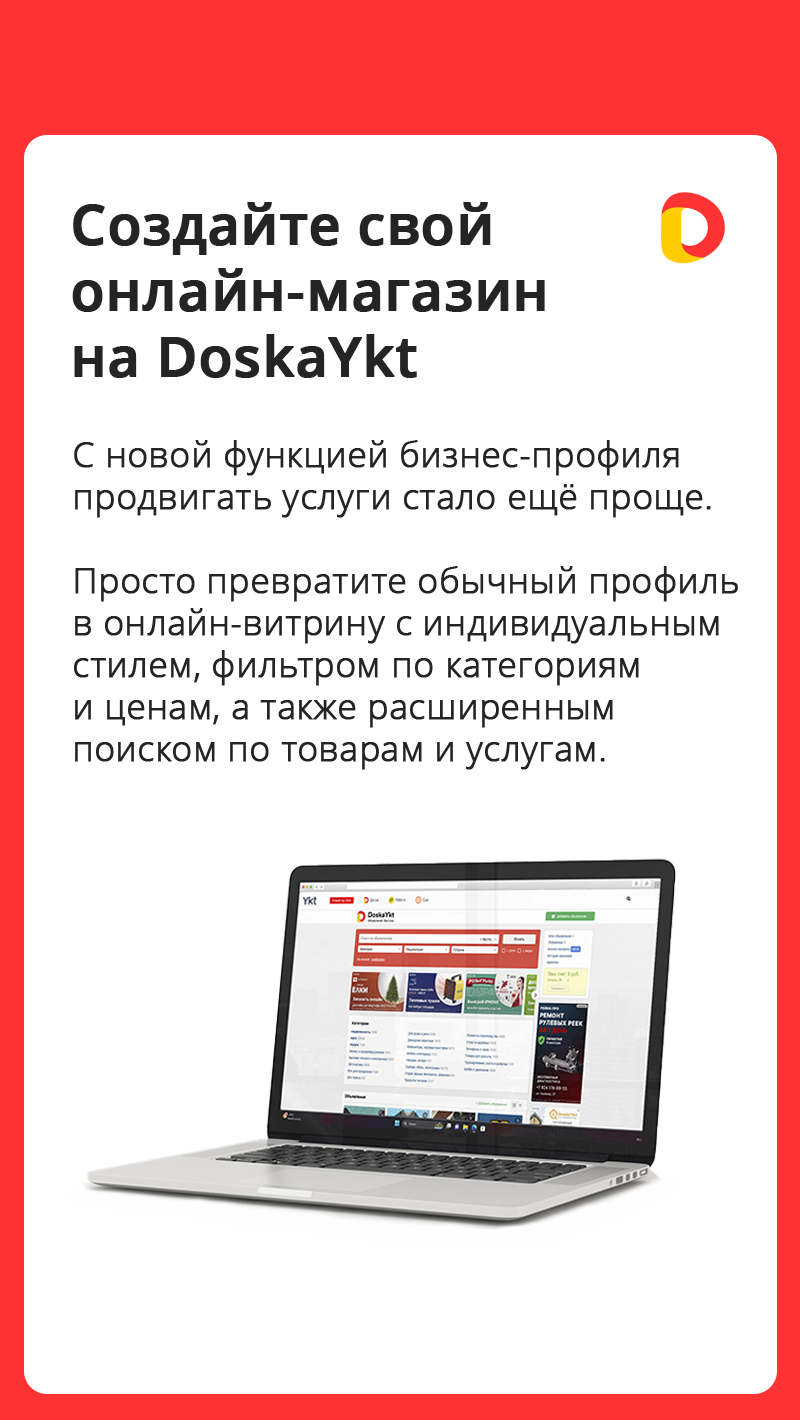
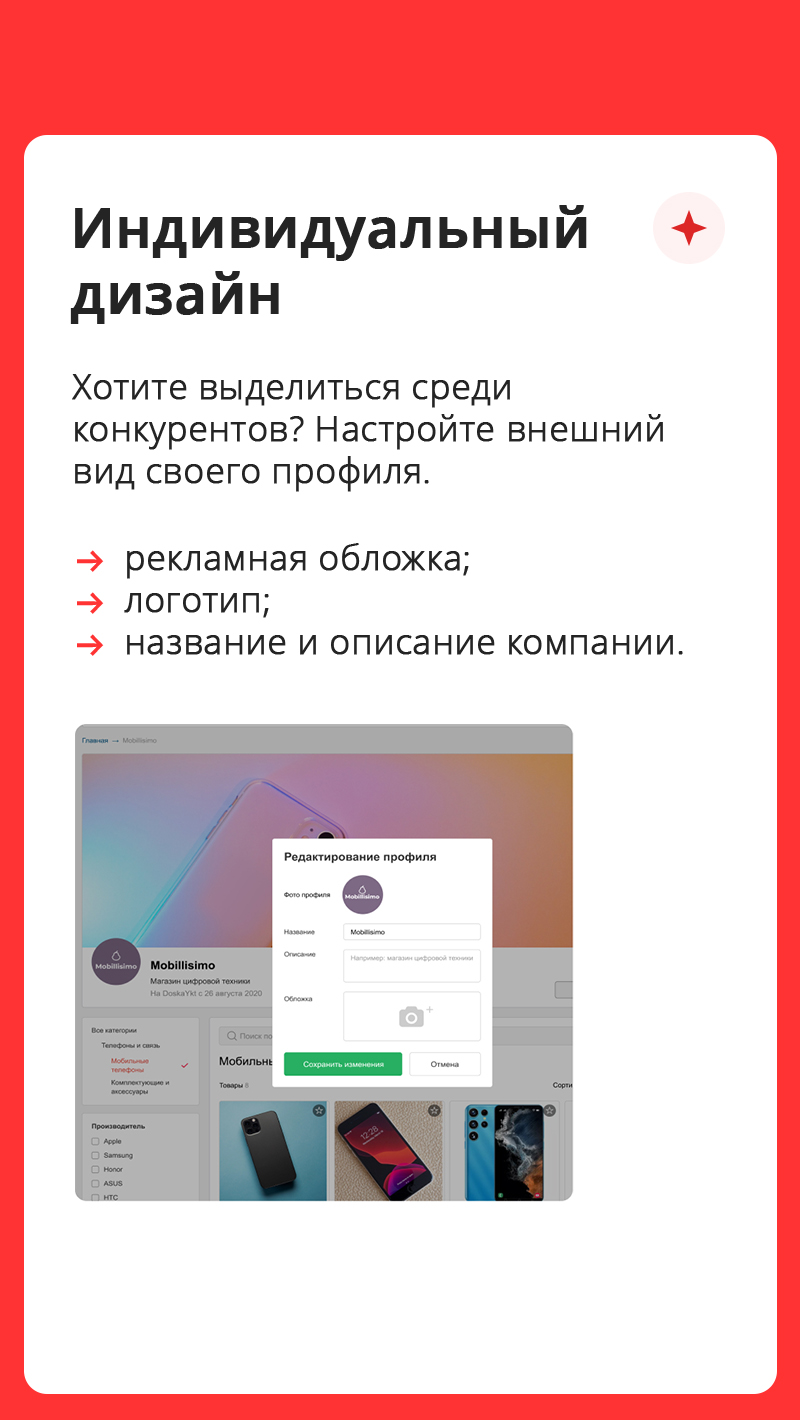
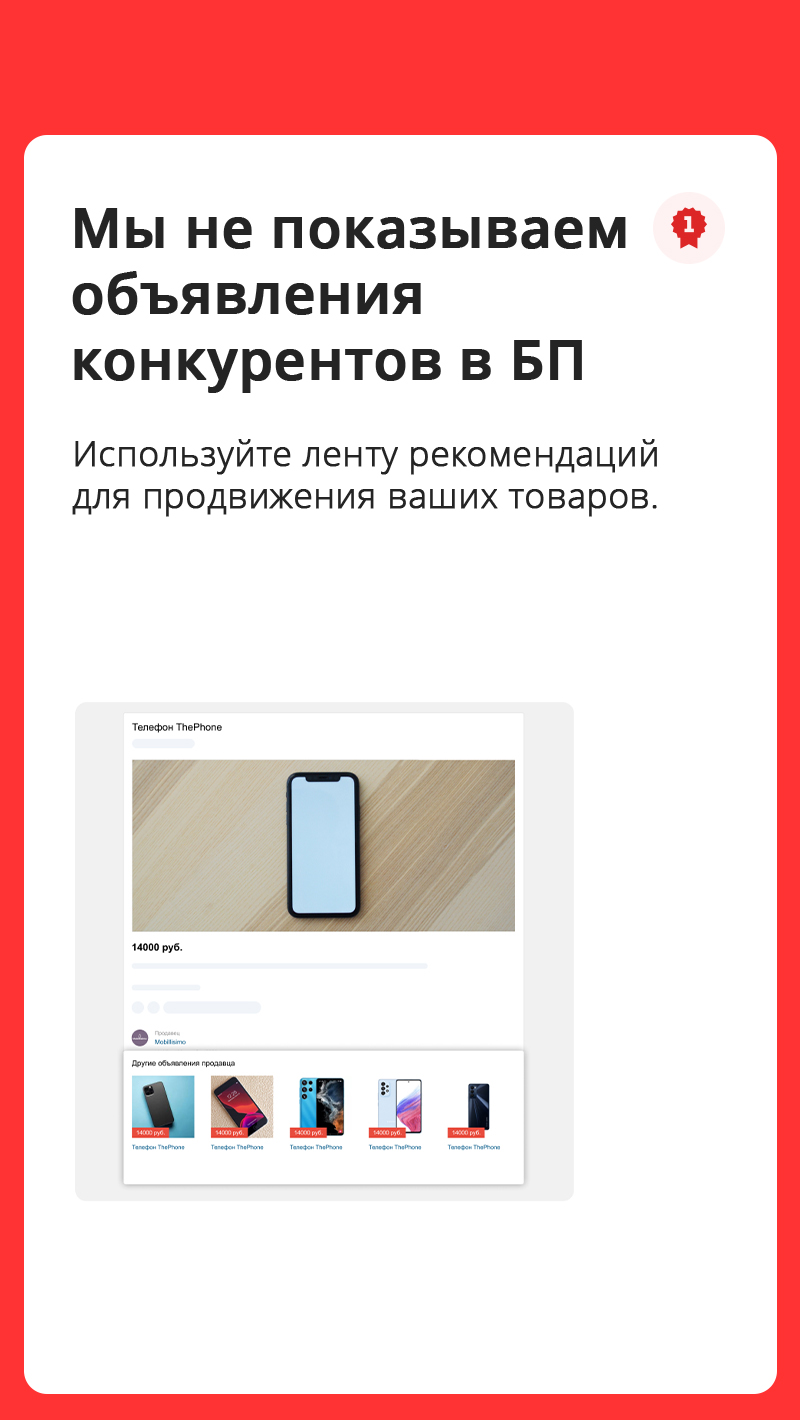
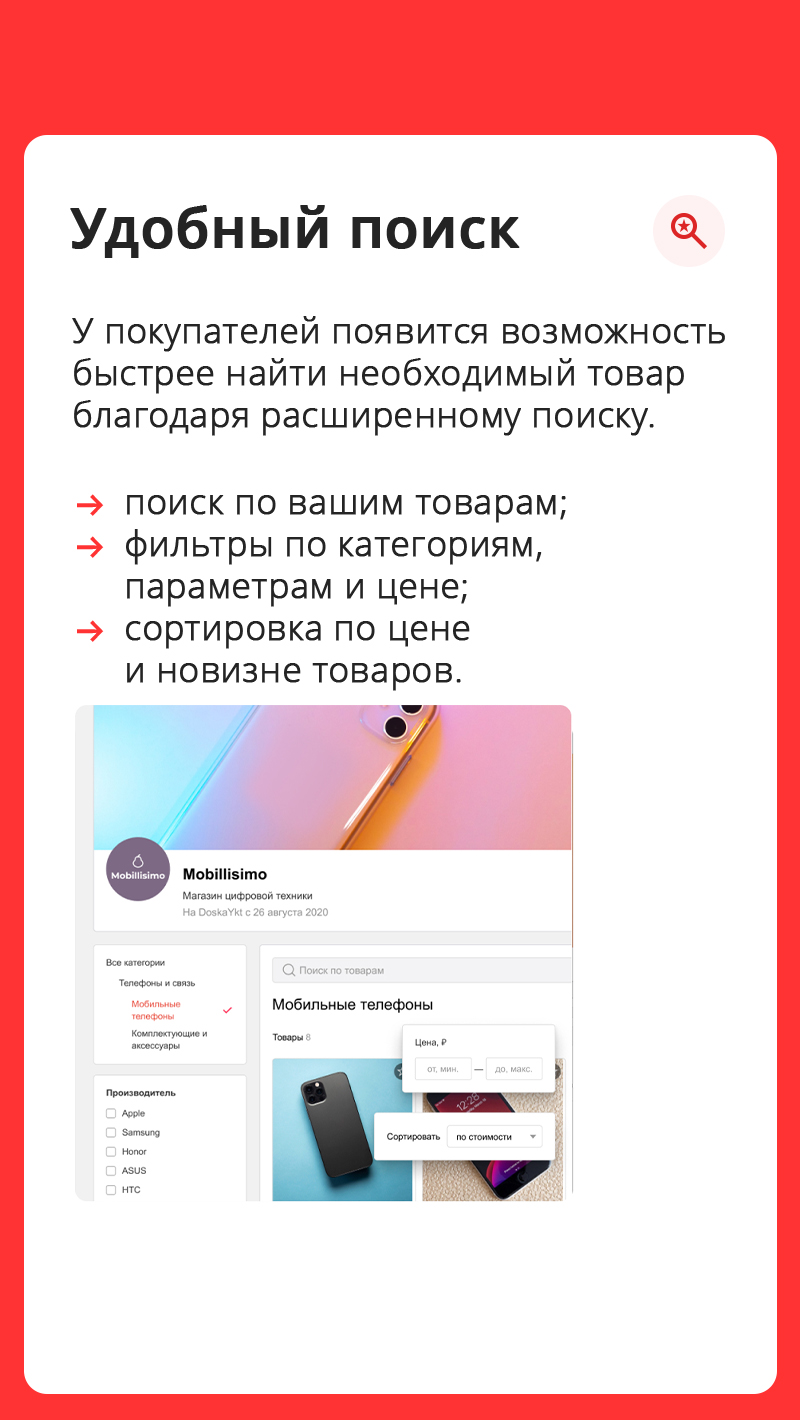
Cвой онлайн-магазин

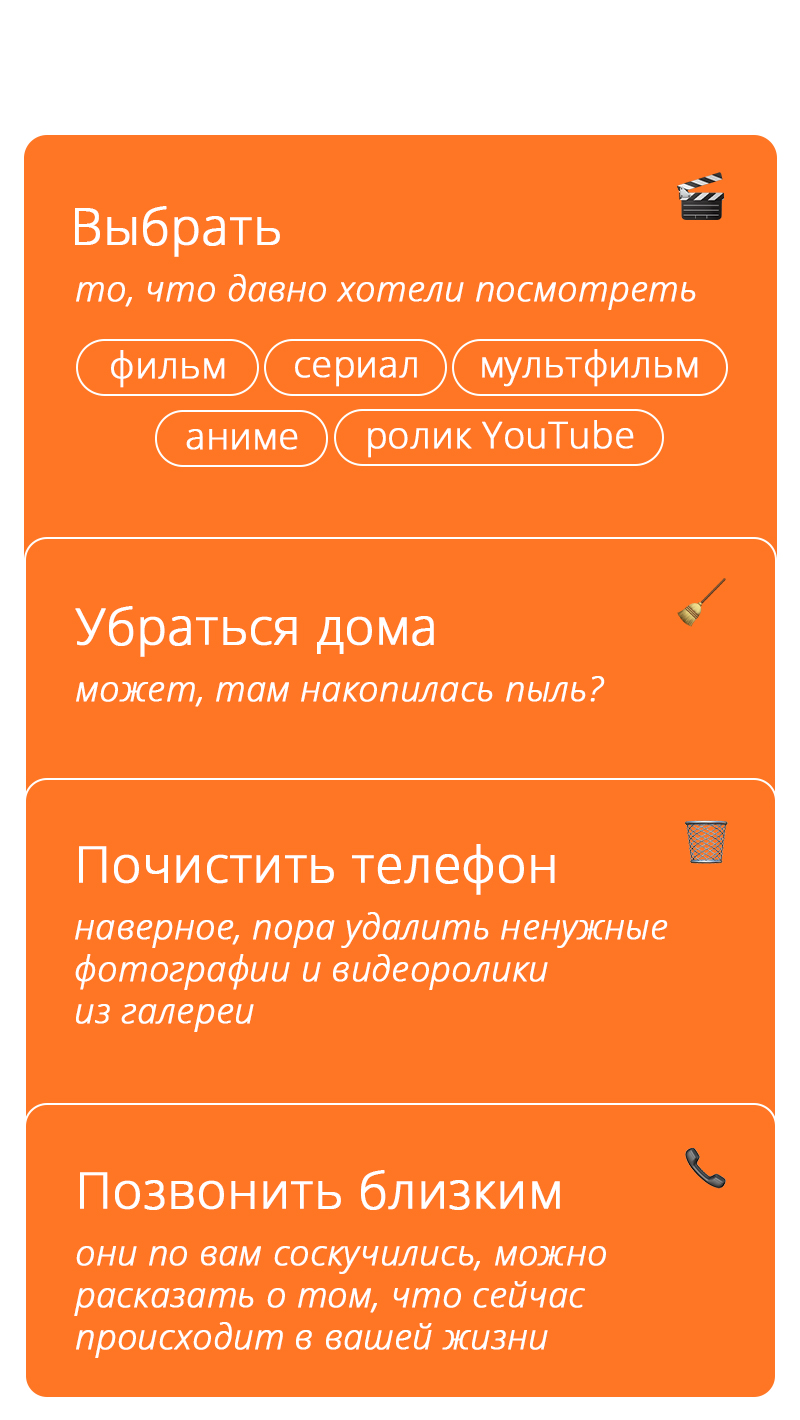
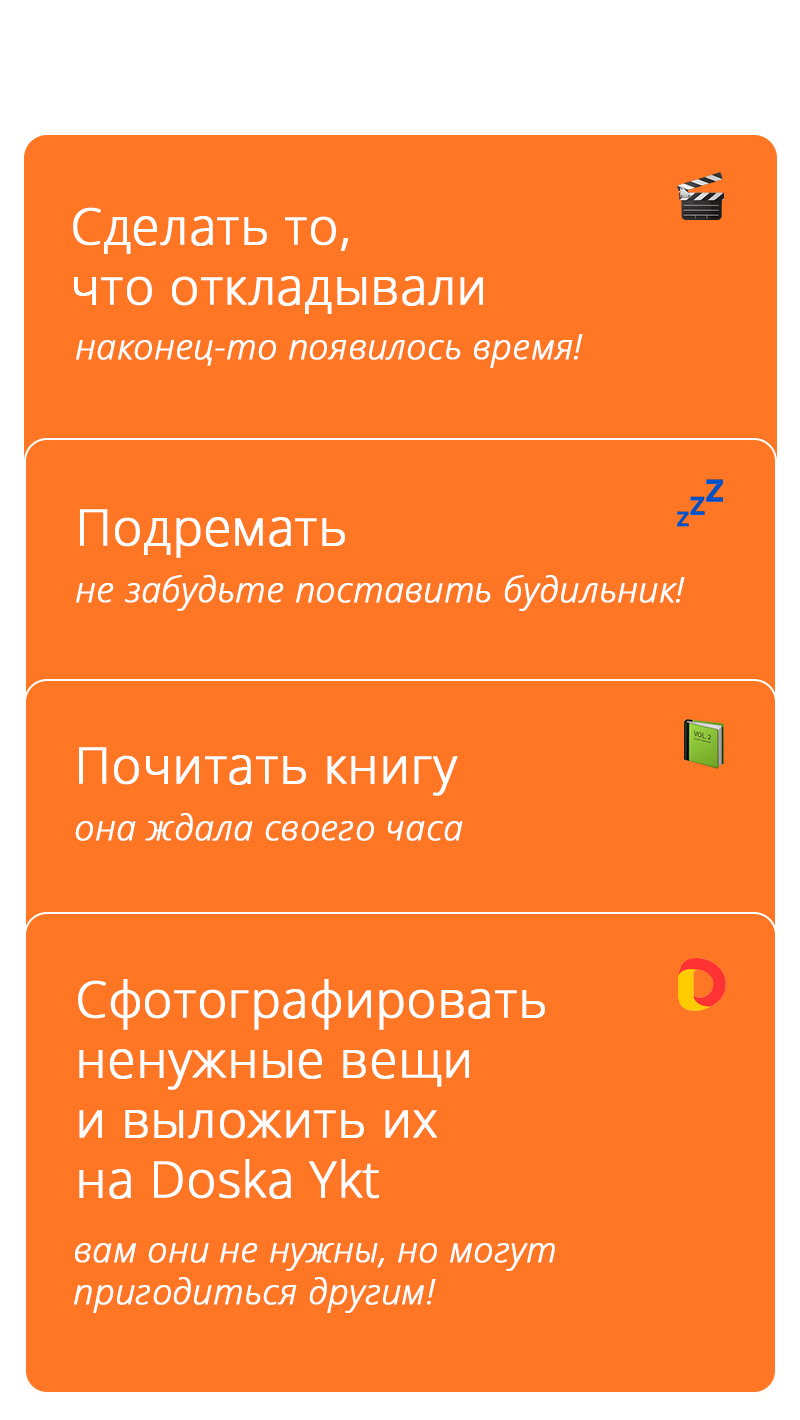
Пока ждёшь доставку

Мы в соцсетях




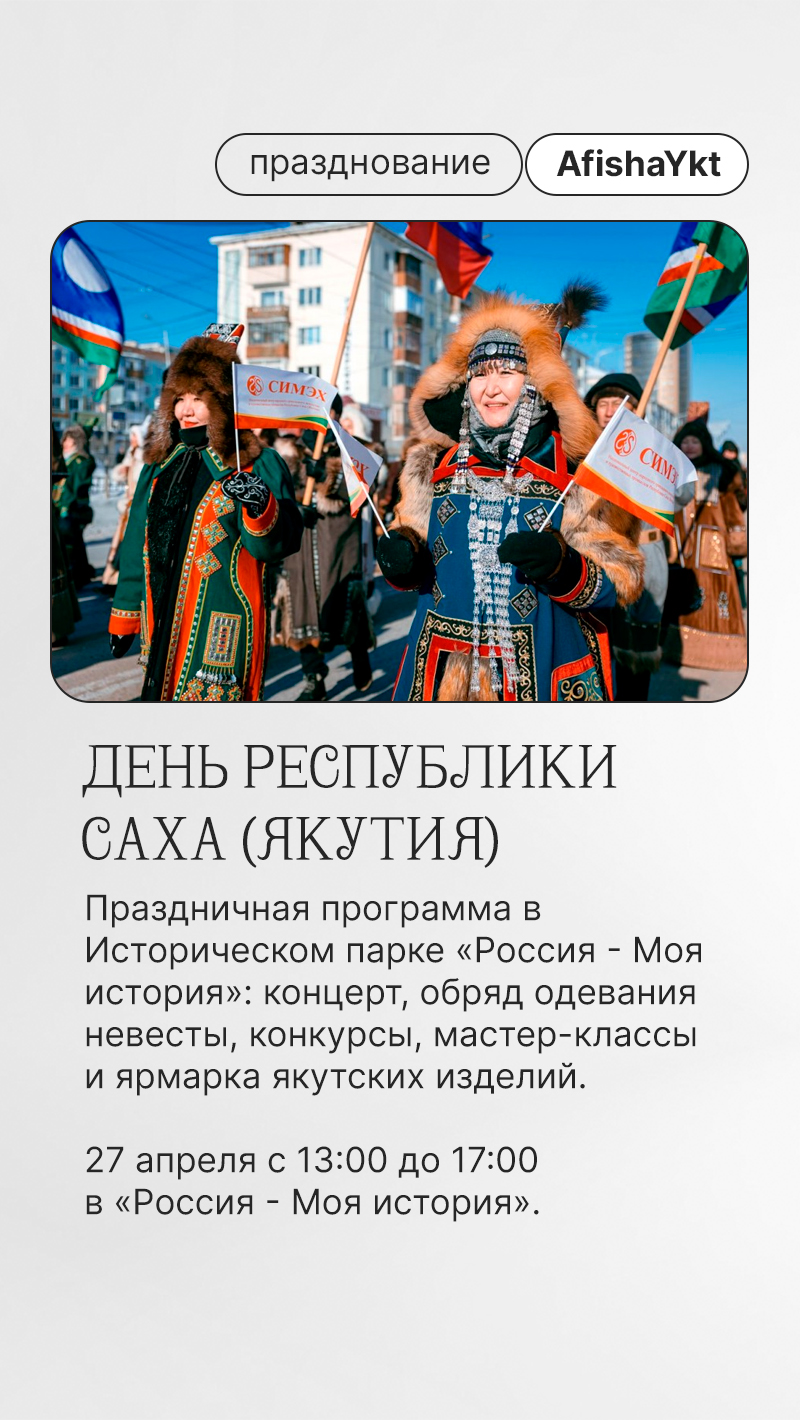











Ykt.Ru











Ykt.Ru













Ykt.Ru















Наши социальные сети